Contributions
Projects

Open Source Vizualizations

Open Source Code
Open Source Data
Projects
GUDMAP/RBK Analytics

Design and manage the Agile group responsible for creating analytics for data stored on the NIH consortiums GUDMAP and RBK. This analytics are not only best-practices pipelines written in Nextflow, but also integrates seamlessly with the consortiums' data-hub, in terms of initiation, and data ingress/egress. It is built with the flexibility to utilize on premises HPC, or a custom built AWS architecture (including many serverless resources) for low-cost, highly-available queueing, compute, and reporting. It can also be easily adapted to almost any running environment using custom configs. The entire pipeline utilizes dockerized micro-services which is orchestrated by Nextflow.
Nextflow in the Cloud
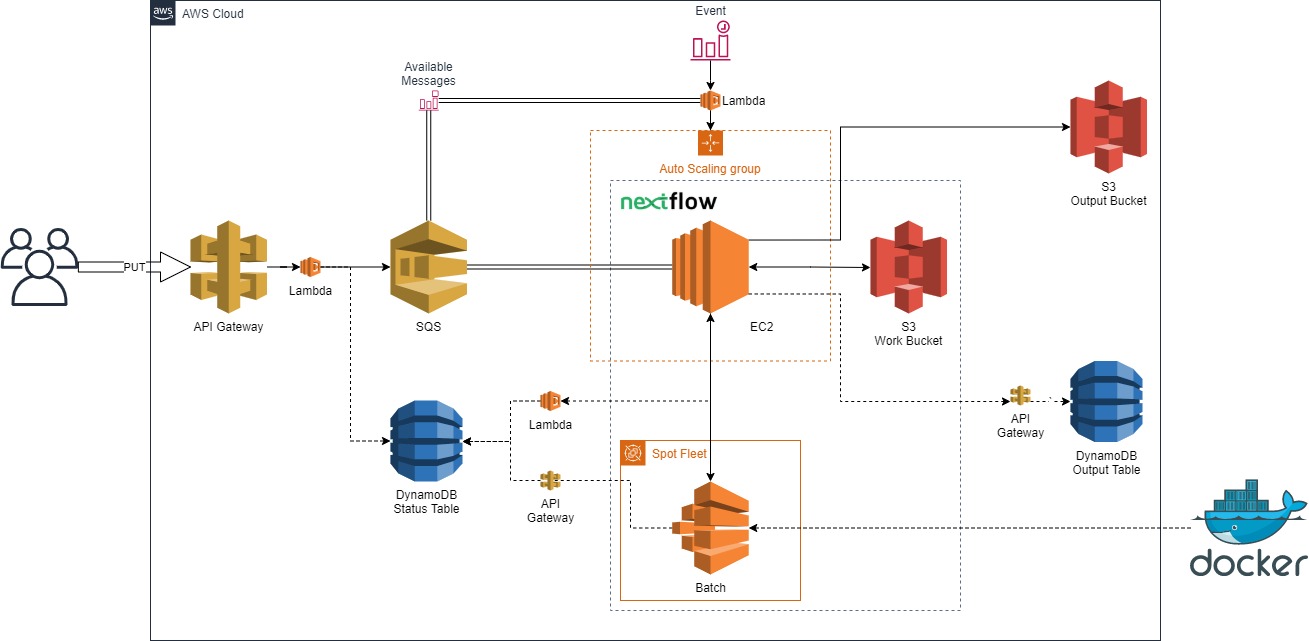
Manage a small agile team to create a proof of concept AWS architecture to run Nextflow pipelines in the cloud. It utilizes low-cost, highly-availability queueing, compute, and storage resources. (NOTE: project decomissioned and the git repository is no longer public, for more information feel free to reach out to [Gervaise Henry](ghenry@gmail.com))
Pipeline Run Tracking
Creating a pipeline run tracking tool for the Bioinformatics Core Facility at UT Southwestern Medical Center. This tool utilizes AWS serverless offerings, including REST-API and a website for run tracking. (NOTE: project decomissioned and the git repository is no longer public, for more information feel free to reach out to [Gervaise Henry](ghenry@gmail.com))
Strand Lab External Website
The Strand Lab website was designed to not only provide a location to share information about the lab, but also provide a means to showcase the lab's data and biorepository. The single-cell data is displayed using CZI's cellxgene visualization tool as well as pre-generated genome-wide visualization of the expression data. The lab has an extensive biorepository of human lower urinary tract biosamples and de-identified clinical data as well as H&E images are available for viewing and filtering on the website. The site is hosted serverlessly on AWS.
Strand Lab Internal Data Website
Developed the internal Strand Lab website and manage the server used explore and share pre-publication single-cell RNA-sequencing data. (NOTE: project is private, for more information feel free to reach out to [Gervaise Henry](ghenry@gmail.com))
BICF Pipelining
Creating analytics for the Bioinformatics Core Facility at UT Southwestern Medical Center, using the pipelining language Nextflow.
Open Source Vizualizations
cellxgene: Single cell analysis of mouse and human prostate reveals novel fibroblasts with specialized distribution and microenvironment interactions

Single-cell RNA-sequencing of adult human prostates and urethras from organ donors, and BPH (glandular and stromal) patients as well as adult mouse lower urinary tracts.
cellxgene: Urethral luminal epithelia are castration-insensitive cells of the proximal prostate
Single-cell RNA-sequencing of adult human prostates and urethras from organ donors, and BPH (glandular) patients as well as adult mouse lower prostates and urethras.
cellxgene: A Cellular Anatomy of the Normal Adult Human Prostate and Prostatic Urethra
Open Source Code
RNA-seq Analytic Pipeline for GUDMAP/RBK
This pipeline conducts end-to-end RNA-seq analysis on replicates stored in the GenitoUrinary Development Molecular Anatomy Project and ReBuilding the Kidney consortium. This pipeline was created in a collaboration between the Strand Lab and Bioinformatics Core Facility (BICF) at UT Southwestern Medical Center.This pipeline conducts end-to-end RNA-seq analysis on replicates stored in the GenitoUrinary Development Molecular Anatomy Project and ReBuilding the Kidney consortium. This pipeline was created in a collaboration between the Strand Lab and Bioinformatics Core Facility (BICF) at UT Southwestern Medical Center.
Strand Lab analysis of single-cell RNA sequencing
This code was used for the single-cell RNA-sequencing analysis in the Strand Lab at UT Southwestern Medical Center
Single-Cell RNA-Seq Image Generation Pipeline for GUDMAP/RBK
This pipeline takes a Seurat object containing single-cell RNA-sequencing data and generates all the plots required to explore the data without querying the data directly. This pipeline was primarily written for uploading the outputs to GenitoUrinary Development Molecular Anatomy Project and ReBuilding the Kidney project. This pipeline was created in a collaboration between the Strand Lab and Bioinformatics Core Facility (BICF) at UT Southwestern Medical Center.
BICF Cellranger mkfastq Analysis Workflow
BICF Cellranger mkfastq Analysis Workflow is a wrapper for the cellranger mkfastq tool from 10x Genomics (which uses Illumina's bcl2fastq). This pipeline, used by the Bioinformatics Core Facility at UT Southwestern, demultiplexes samples from 10x Genomics single-cell libraries into fastqs. FastQC is run on the results all reports are collated with the MultiQC tool. This pipeline is primarily used with a SLURM cluster on BioHPC, but it should be able to run on any system that Nextflow supports. Additionally, this pipeline is designed to work using a simple web interface.
BICF Cellranger count Analysis Workflow
BICF Cellranger count Analysis Workflow is a wrapper for the cellranger count tool from 10x Genomics. This pipeline, used by the Bioinformatics Core Facility at UT Southwestern, takes fastq files from 10x Genomics single-cell gene expression libraries and passes them to cellranger count, managing parallelization of multiple runs, as well as, aggregation as appropriate. This pipeline is primarily used with a SLURM cluster on BioHPC, but it should be able to run on any system that Nextflow supports. Additionally, this pipeline is designed to work using a simple web interface.
Determining cellular heterogeneity in the human prostate with single-cell RNA sequencing
This is the code which was used for the single-cell RNA-sequencing analysis published in Cell Reports 2018 and used the data in GenitoUrinary Development Molecular Anatomy Project
Open Source Data
GEO: Human prostate luminal epithelia adopt a club-like state in response to low androgen levels

Spatial transcriptomics of treatment naive and 5-alpha reductase inhibitior treated human BPH tissue
GEO: Single cell RNA-sequencing of verumontanum region and peripheral zone of normal human prostate
Dissected verumontanum region and peripheral zone region from a normal human prostate sample were digested into single-cell suspensions, cells were subject to single-cell RNA-sequencing using the 10x Genomics platform
GEO: Bulk RNA-sequencing of fibroblasts isolated by FACS from normal human prostate
Differential expression of prostatic fibroblasts were determined using RNA-sequencing of FACS sorted fibroblasts from normal human prostate transition and peripheral.
GEO: Single-cell RNA-sequencing of adult human normal and BPH (glandular and stroma) prostates and urethra
Single-cell RNA-sequencing of adult human prostates and urethras from organ donors, and BPH (glandular and stromal) patients.
GEO: Single-cell RNA-sequencing of adult mouse lower urinary tracts v2
GUDMAP: Single cell analysis of mouse and human prostate reveals novel fibroblasts with specialized distribution and microenvironment interactions

Sequencing data relating to the in preparation publication: 'Single cell analysis of mouse and human prostate reveals novel fibroblasts with specialized distribution and microenvironment interactions'
GUDMAP: Urethral luminal epithelia are castration-insensitive cells of the proximal prostate
Figures and data relating to the 2020 Prostate paper titled “Urethral luminal epithelia are castration‐insensitive cells of the proximal prostate”. This study utilizes single-cell RNA-sequencing (scRNA-Seq) on whole prostate tissues from healthy organ donors, basal cell hyperplasia patients, as well as mouse prostate and urethral tissues.
GEO: Single-cell RNA-sequencing of adult mouse lower urinary tracts
GEO: Single-cell RNA-sequencing of adult human prostates and urethra (normal and diseased)
Single-cell RNA-sequencing of adult human prostates from organ donors, and BPH (glandular) patients.
FLOWRepository: OMIP-040: Optimized gating of human prostate cellular subpopulations

Epithelia, stoma and leukocytes can be isolated using CD45 and CD326. Epithelia are CD326Pos/CD45Neg, leukocytes are CD45Pos/CD26Neg, while stroma are double negative for these markers. Stroma can be separated into endothelia and fibromuscular stroma with CD31. Endothelia are CD31Pos while fibromuscular stroma are CD31Neg. Epithelia can also be divided into basal and luminal epithelia using the basal marker CD271 and the luminal marker CD26.
GEO: Bulk RNA-sequencing of cell types isolated by FACS from normal human prostates
Differential expression of prostatic cell types were determined using RNA-sequencing of FACS sorted cell types from normal human prostates. The cell types sequenced were basal epithelia, luminal epithelia, a heterogeneous fibromuscular stroma and a poorly understood 'other' epithelial population.
GEO: Single Cell RNA-sequencing of cell types isolated by FACS from normal human prostates
Single-cell RNA-sequencing was conducted on specific cell types and heterogeneous viable cells (sorted by FACS) in order to determine a cellular atlas of the normal human prostate.
GUDMAP: Determining cellular heterogeneity in the human prostate with single-cell RNA sequencing
Figures and data relating to the 2018 Cell Reports paper titled “A Cellular Anatomy of the Normal Adult Human Prostate and Prostatic Urethra”. This study utilizes single-cell RNA-sequencing (scRNA-Seq) on whole prostate tissue and FACS sorted cell populations from healthy organ donors to predict the cellular composition of the normal human prostate.